Anemia – objawy, przyczyny i rozpoznawanie niedokrwistości
Dowiedz się więcej

Choroba Rendu-Oslera-Webera (dziedziczna krwotoczna teleangiektazja (HHT)) to choroba uwarunkowana genetycznie charakteryzująca się nieprawidłową budową i funkcjonowaniem naczyń krwionośnych w obrębie skóry, błon śluzowych i narządów wewnętrznych. Do typowych objawów zalicza się krwawienia z nosa, teleangiektazję skórne (poszerzone drobne naczynia krwionośne, „pajączki”), krwawienia z przewodu pokarmowego oraz malformacje tętniczo-żylne (nieprawidłowa budowa naczyń krwionośnych). Przyczyną choroby są mutacje genowe. W poniższym artykule charakteryzujemy przyczyny i objawy choroby Rendu-Oslera-Webera oraz opisujemy, jak wygląda proces diagnostyczny.

Choroba Rendu-Oslera-Webera jest schorzeniem dziedziczonym autosomalnie dominująco, oznacza to, że do wystąpienia objawów choroby wystarczy posiadanie tylko jednej kopii uszkodzonego genu odziedziczonej od matki lub ojca. Osoba dotknięta chorobą ma 50% szans na przekazanie zmutowanego genu swojemu potomstwu. Częstość występowania choroby nie jest dokładnie poznana, ale szacuje się, że wynosi ona około od 1:5000 do 1:8000. Do tej pory opisano kilka mutacji, które są odpowiedzialne za wystąpienie symptomów chorobowych. W większości przypadków mutacja warunkująca rozwój obrazu klinicznego dziedzicznej krwotocznej teleangiektazji dotyczy genu ENG, zlokalizowanego na chromosomie 9, genu ACVRL1 zlokalizowanego na chromosomie 12 lub genu BMPR2 zlokalizowanego na chromosomie 2. W nielicznych przypadkach przyczyną choroby są mutacje genu HHT3 (chromosom 5), HHT4 (chromosom 7) lub genu GGF2 (chromosom 10).
Cechą charakterystyczną zespołu Rendu-Oslera-Webera jest występowanie licznych malformacji tętniczo-żylnych (AVM) na powierzchni skóry, błon śluzowych lub narządów wewnętrznych. Stopień nasilenia objawów klinicznych choroby wzrasta wraz z wiekiem. Początkowo pojawiają się nawracające krwawienia z nosa a w późniejszym wieku charakterystyczne dla HHT teleangiektazje twarzy, gardła i rąk. Na późniejszym etapie choroby malformacje naczyniowe mogą obejmować przewód pokarmowy, płuca, mózg, rdzeń kręgowy powodując krwotoki wewnątrzczaszkowe, ropnie, duszność wysiłkową, niewydolność serca, przewlekłe niedotlenienie, krwawienia z przewodu pokarmowego lub niewydolność wątroby.
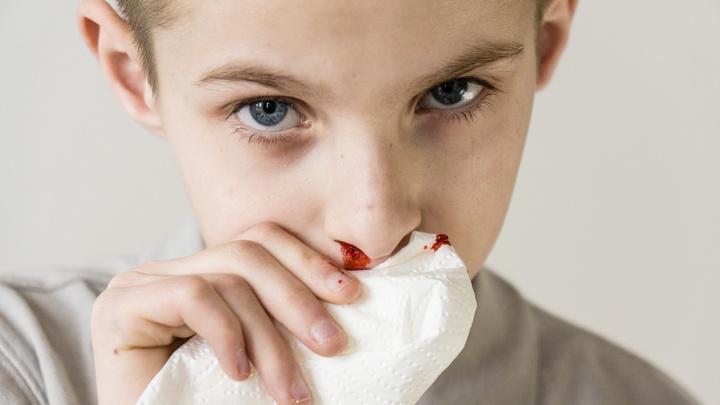
U około 95% pacjentów obserwuje się występowanie nawracających krwawień z nosa przed ukończeniem 20. roku życia. Krwawienia z nosa nasilają się wraz z wiekiem chorego i mogą prowadzić do niedokrwistości z niedoboru żelaza. Mogą występować spontanicznie lub w wyniku działania czynnika drażniącego np. urazu mechanicznego, lub suchości błon śluzowych.
U większości pacjentów obserwuje się występowanie licznych teleangiektazji – „pajączków” w obrębie twarzy, ust, palców lub pod paznokciami. Teleangiektazje bledną pod wpływem nacisku na skórę, co pozwala na odróżnienie ich od wybroczyn. Ilość i wielkość tych zmian zwiększa się wraz z wiekiem pacjentów.
U około 15-50% chorych występują malformacje tętniczo-żylne w obrębie płuc, najczęściej w okolicach dolnych płatów płucnych. Zmiany te mogą początkowo przebiegać bezobjawowo, jednak w niektórych przypadkach prowadzą do duszności, niedotlenienia i nadciśnienia płucnego. Pacjenci najczęściej skarżą się na duszności pojawiające się w trakcie wysiłku fizycznego. W rzadkich przypadkach u chorych może występować krwioplucie spowodowane pęknięciem kruchych, zniekształconych naczyń krwionośnych lub zagrażający życiu krwotok płucny.
Teleangiektazje w obrębie przewodu pokarmowego występują głównie w żołądku i jelicie cienkim. Najczęściej mają charakter bezobjawowy, ale u około 45% pacjentów może wystąpić krwotok żołądkowo-jelitowy. Krwawienia z przewodu pokarmowego pojawiają się zazwyczaj około 50-60. roku życia, lecz u części chorych mogą nie wystąpić wcale. Malformacje żylno-tętnicze w wielu przypadkach dotyczą również wątroby. Konsekwencjami tych zmian jest nadciśnienie wrotne, żylaki przełyku, zastoinowa niewydolność serca lub nieprawidłowe funkcjonowanie dróg żółciowych. W skrajnych przypadkach może wystąpić niewydolność wątroby i konieczność wykonania przeszczepu.
Zmiany neurologiczne u pacjentów z chorobą Rendu-Oslera-Webera mogą być następstwem zmian malformacyjnych powstałych w obrębie płuc lub układu nerwowego. Zalicza się do nich udary niedokrwienne, ropnie mózgu, ataki niedokrwienne.
📌 Sprawdź Pierwotne zapalenie dróg żółciowych – co to za choroba? Objawy, przyczyny, rozpoznawanie
Diagnoza choroby Rendu-Oslera-Webera opiera się na kryteriach klinicznych, wynikach badań genetycznych oraz wynikach badań obrazowych. Do kryteriów klinicznych zalicza się występowanie krwawień z nosa; licznych teleangiektazji w charakterystycznych dla choroby miejscach (usta, twarz, palce, nos, jama ustna); obecność zmian trzewnych – malformacji żylno-tętniczych w obrębie płuc, mózgu, wątroby, rdzenia kręgowego, żołądka, jelit oraz obciążenia rodzinnego, czyli występowania choroby u krewnych pierwszego stopnia. Diagnoza uważana jest za pewną, jeśli spełnione są co najmniej trzy kryteria. Niestety wiele przypadków choroby nie jest zdiagnozowane ze względu na fakt, że poszczególne symptomy charakterystyczne dla choroby Rendu-Oslera-Webera mogą pojawiać i nasilać się w różnym wieku, co sprawia szczególne trudności diagnostyczne u osób młodych. Istotną rolę w postawieniu odpowiedniego rozpoznania odgrywają badania genetyczne sprawdzające obecność mutacji genowych odpowiedzialnych za rozwój choroby, czyli mutacji genów ENG, ACVRL1 HHT3, HHT4, BMPR2 lub GGF2.