Atopowe zapalenie skóry (ICD-10: L20) – przyczyny, objawy, diagnostyka
Dowiedz się więcej

Skóra jako największy i jeden z najbardziej złożonych narządów organizmu, pełni funkcję wyspecjalizowanej, wielofunkcyjnej bariery ochronnej. Stanowi wytrzymałą i elastyczną powłokę, która odgrywa kluczową rolę w utrzymaniu homeostazy ustroju. Ponadto, jest wysoko unerwioną strukturą czuciową, umożliwiającą percepcję bodźców mechanicznych, termicznych oraz bólowych. Jej integralność strukturalna ma zatem zasadnicze znaczenie dla prawidłowego funkcjonowania całego organizmu. W świetle tej wiedzy szczególnie istotne staje się pytanie o konsekwencje kliniczne wrodzonych patologii, które radykalnie naruszają tę integralność. Przykładem takiej jednostki chorobowej jest pęcherzowe oddzielanie się naskórka (EB), w przebiegu której nawet minimalny uraz mechaniczny prowadzi do powstawania rozległych pęcherzy oraz ran. Ze względu na wyjątkową delikatność i podatność na uszkodzenia, skóra pacjentów z EB bywa porównywana do skrzydeł motyla, co znajduje swoje odzwierciedlenie w potocznej nazwie tego schorzenia – „choroba Dzieci-Motyli”. EB uznawana jest za jedno z najboleśniejszych schorzeń na świecie, a przewlekły ból związany z ciągłym gojeniem się ran stanowi stały element życia chorych.
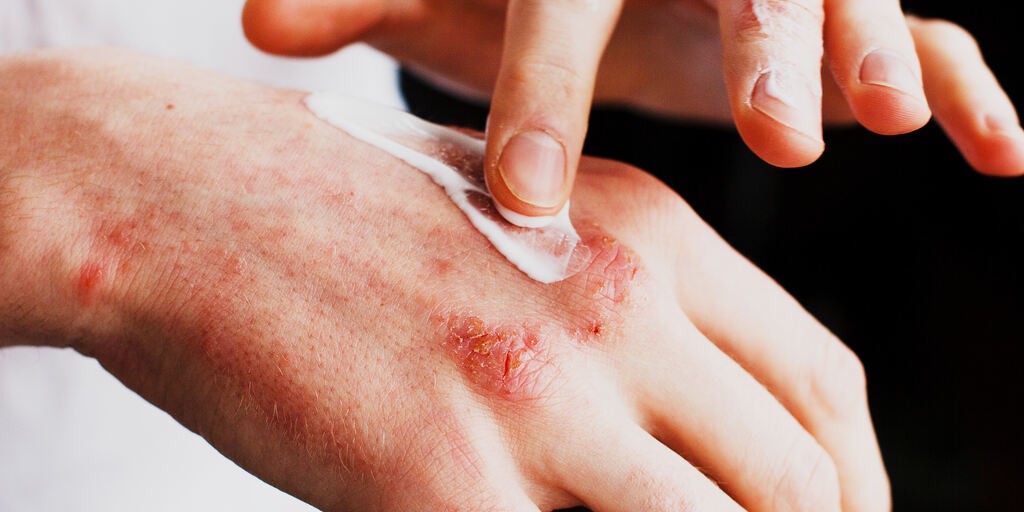
Epidermolysis Bullosa (EB) znane w nomenklaturze medycznej jako pęcherzowe oddzielanie się naskórka, stanowi heterogenną grupę rzadkich genodermatoz, których wspólnym patomechanizmem jest genetycznie uwarunkowana kruchość strukturalna skóry, prowadząca do powstawania pęcherzy i nadżerek w odpowiedzi na najmniejsze urazy mechaniczne. W przebiegu tego schorzenia nawet codzienne zabiegi pielęgnacyjne, ucisk czy tarcie mogą doprowadzić do powstawania przewlekłych ran. Choroba charakteryzuje się niezwykle zróżnicowanym i często ciężkim spektrum objawów klinicznych. Pęcherzowe oddzielanie się naskórka jest schorzeniem o podłożu genetycznym, obecnym od urodzenia, choć jego manifestacja kliniczna może ujawniać się dopiero w późniejszym okresie życia.
Z uwagi na rzadki charakter schorzenia, większość personelu medycznego nie posiada doświadczenia niezbędnego do jego kompleksowego leczenia. Szacuje się, że na świecie żyje około 500 000 osób z EB, a chorobę tą rozpoznaje się średnio u 1 na 30 000 żywych urodzeń. Warto podkreślić, że jej występowanie jest niezależne od płci i w równej mierze rozkłada się pomiędzy różnymi grupami etnicznymi, nie wykazując przy tym żadnych geograficznych preferencji. Według dostępnych danych w Polsce choruje około 500 osób. Dotychczas sklasyfikowano 27 odrębnych fenotypów EB, odzwierciedlających jego zróżnicowane przejawy kliniczne oraz ponad 100 różnorodnych genotypów wynikających z mutacji
w poszczególnych loci genowych. Aktualna klasyfikacja, oparta na triadzie kryteriów: miejscu występowania pęcherza w strukturze skóry (poziom rozszczepu), obrazie klinicznym oraz zidentyfikowanej mutacji genetycznej, wyodrębnia cztery fundamentalne typy choroby:
Każdy z wymienionych typów choroby ma unikalną etiologię molekularną, wynikającą
z mutacji w genach kodujących odrębne białka strukturalne. Kluczowym badaniem diagnostycznym pozwalającym na klasyfikację głównych typów EB jest analiza histologiczna wycinka skóry, pobranego z miejsca tworzenia się pęcherza. Głównym celem tego badania jest precyzyjne zlokalizowanie strukturalnego poziomu, na którym dochodzi do rozszczepu i akumulacji płynu w obrębie błony podstawnej, stanowiącej granicę pomiędzy naskórkiem
a skórą właściwą. Na podstawie obrazu mikroskopowego możliwe jest ustalenie rozpoznania:
W obrębie typu łączącego wyróżnia się ponadto szczególnie ciężką odmianę, tzw. typ Herlitz. Charakteryzuje się on rozległym tworzeniem się pęcherzy już od pierwszych dni życia oraz szybkim rozwojem nadżerek obejmujących zarówno skórę, jak i błony śluzowe. Przebieg choroby jest gwałtowny i wiąże się z wysokim ryzykiem powikłań ogólnoustrojowych. Ze względu na ciężkość objawów klinicznych rokowanie w tej postaci jest bardzo niekorzystne, a większość chorych nie przeżywa wieku niemowlęcego.
Objawy pęcherzowego oddzielania się naskórka charakteryzują się dużą zmiennością kliniczną i są uzależnione od typu oraz ciężkości choroby. Przebieg kliniczny EB może przybierać różne formy, od postaci ograniczonych z nielicznymi i szybko gojącymi się zmianami, aż po ciężkie, rozległe i przewlekłe owrzodzenia obejmujące znaczną część ciała. Podstawowym i najbardziej charakterystycznym objawem EB jest skłonność do powstania pęcherzy, nadżerek i ran na skórze oraz błonach śluzowych w odpowiedzi na niewielkie urazy mechaniczne, tarcie lub ucisk. Zmianom skórnym często towarzyszy świąd, ból, przewlekły stan zapalny oraz zaburzenie procesu gojenia, które prowadzi do bliznowacenia, zaników skóry i deformacji anatomicznych, zwłaszcza w cięższych postaciach choroby. U wielu pacjentów obserwuje się również zajęcie błon śluzowych układu oddechowego, moczowo-płciowego, jamy ustnej, przełyku i innych odcinków przewodu pokarmowego. Skutkuje to trudnościami w przyjmowaniu pokarmów, niedożywieniem oraz zaburzeniem wzrastania. Dodatkowo w literaturze medycznej opisywane są przypadki występowania zmian paznokciowych, utraty owłosienia, przewlekłego zakażenia skóry oraz wtórnej anemii. Te współistniejące z EB schorzenia znacząco obniżają jakość życia chorych i stanowią istotne wyzwanie kliniczne w długoterminowej opiece nad pacjentami cierpiącymi na tę chorobę.
Pęcherzowe oddzielanie się naskórka to choroba, której istotą są zaburzenia w obrębie struktur odpowiedzialnych za stabilność i wytrzymałość skóry. Mutacje dotyczą genów kodujących białka pełniące kluczową funkcję w utrzymaniu spójności pomiędzy naskórkiem a skórą właściwą. U osób zdrowych białka te tworzą złożony system połączeń komórkowych, umożliwiając skórze skuteczną ochronę przed urazami mechanicznymi. W przebiegu choroby EB dochodzi jednak do osłabienia tych mechanizmów, przez co odporność skóry na działanie nawet niewielkich sił zewnętrznych zostaje znacząco obniżona. Patomechanizm schorzenia jest ściśle skorelowany z konkretnym genetycznym defektem molekularnym.
Dziedziczenie może odbywać się w sposób autosomalny dominujący lub autosomalny recesywny. W przypadku dziedziczenia dominującego do ekspresji choroby wystarczy obecność jednej zmutowanej kopii genu, która zazwyczaj pochodzi od rodzica z pełnoobjawową postacią EB. Dziedziczenie recesywne wymaga odziedziczenia dwóch wadliwych alleli, przy czym rodzice są najczęściej bezobjawowymi nosicielami tych mutacji. Istnieje również możliwość wystąpienia mutacji spontanicznej de novo, bez wcześniejszego występowania choroby w linii rodzinnej. Ostateczny fenotyp kliniczny, obejmujący głębokość powstawania pęcherza oraz ciężkość przebiegu, jest determinowany przez rodzaj zaangażowanego genu i charakter mutacji. Defekty te dotyczą białek strukturalnych, takich jak keratyny w obrębie tonofilamentów, składników hemidesmosomów (np. kolagen typu XVII, białko BP180) lub białek kotwiczących włókna (kolagen typu VII). Niedobór, nieprawidłowa budowa lub całkowity brak syntezy tych białek prowadzi do destabilizacji połączenia skórno-naskórkowego. W efekcie nawet minimalne siły ścinające, takie jak tarcie lub ucisk, powodują rozszczepienie na poziomie histologicznym, manifestujące się powstawaniem wiotkich lub napiętych pęcherzy, a następnie bolesnych nadżerek i trudno gojących się ran.
Postępowanie terapeutyczne w EB ma obecnie charakter głównie objawowy i pielęgnacyjny, a jego celem jest zapobieganie powstawaniu nowych zmian skórnych oraz optymalizacja procesu gojenia się istniejących już ran. Podstawą leczenia jest kompleksowa ochrona skóry przed urazami mechanicznymi, realizowana poprzez systematyczne stosowanie specjalistycznych, nieprzylegających opatrunków, które ograniczają tarcie, ucisk oraz ryzyko wtórnego uszkodzenia naskórka. Kluczowe znaczenie ma regularna, często wielokrotnie wykonywana w ciągu doby, zmiana opatrunków z wykorzystaniem preparatów antyseptycznych i odpowiednich środków pielęgnacyjnych. Takie działania pozwalają zmniejszyć ryzyko wtórnych zakażeń bakteryjnych i przyspieszyć proces regeneracji tkanek.
W cięższych postaciach choroby leczenie obejmuje również farmakoterapię infekcji, leczenie bólu oraz interwencje chirurgiczne o charakterze korekcyjnym, takie jak usuwanie zrostów palców czy zabiegi rekonstrukcyjne przełyku. U wybranych pacjentów stosuje się także stworzone za pomocą bioinżynierii substytuty skóry, które wspomagają leczenie rozległych i przewlekłych ubytków. Istotnym osiągnięciem współczesnej medycyny jest wprowadzenie miejscowej terapii genowej, zatwierdzonej do stosowania w krajach Unii Europejskiej w leczeniu dystroficznej postaci EB, która polega na dostarczaniu funkcjonalnej kopii genu COL7A1 do komórek rany za pośrednictwem niezdolnego do replikacji wektora wirusa opryszczki pospolitej (HSV-1). Prowadzi to do zwiększenia syntezy kolagenu typu VII i poprawy gojenia się ran. Choć terapia ta nie eliminuje przyczyny choroby i wymaga powtarzalnych aplikacji, wykazano jej istotny wpływ na skrócenie czasu gojenia oraz redukcję przewlekłych owrzodzeń. Z uwagi na genetycznych charakter EB leczenie koncentruje się przede wszystkim na łagodzeniu objawów, zapobieganiu powikłaniom i poprawie jakości życia pacjentów, przy czym kluczowe znaczenie ma indywidualnie dostosowana, wielospecjalistyczna opieka oraz zapewnienie dostępu do refundowanych materiałów opatrunkowych i medycznego wsparcia długoterminowego.
Oczekiwana długość życia pacjentów z pęcherzowym oddzielaniem się naskórka (EB) jest ściśle uzależniona od konkretnego podtypu choroby. W łagodnych, ograniczonych postaciach EB simplex prognoza przeżycia jest zazwyczaj korzystna i zbliżona do populacji ogólnej. W postaciach ciężkich, takich jak uogólniona recesywna dystroficzna postać EB (ang. Recessive Dystrophic Epidermolysis Bullosa, RDEB) czy letalne warianty EB łączącej, długość życia ulega znacznemu skróceniu. Pacjenci z najbardziej agresywnymi fenotypami rzadko przeżywają wczesne dzieciństwo. Osoby z RDEB, mimo że często osiągają wiek dorosły, pozostają narażone na przedwczesną śmierć, zwykle w trzeciej lub czwartej dekadzie życia, najczęściej wskutek przerzutów raka kolczystokomórkowego skóry, który rozwija się jako powikłanie przewlekłych, trudno gojących się owrzodzeń. Wprowadzenie nowoczesnej, wielospecjalistycznej opieki medycznej, skoncentrowanej na profilaktyce powikłań i leczeniu wspomagającym, może istotnie poprawiać rokowanie w tej grupie pacjentów.
Mgr Karolina Hejnar
Przewidywana długość życia pacjentów z chorobą pęcherzową (EB) jest ściśle uzależniona od rodzaju i nasilenia postaci choroby. W przypadkach łagodnych możliwe jest osiągnięcie wieku zbliżonego do przeciętnej długości życia populacji ogólnej. W ciężkich wariantach, takich jak dystroficzna EB w postaci ciężkiej, średnia długość życia jest istotnie skrócona i zwykle nie przekracza trzeciej dekady życia, przy czym wdrożenie kompleksowej opieki medycznej i profilaktyki powikłań może znacząco modyfikować rokowanie.
Epidermolysis bullosa (EB), czyli pęcherzowe oddzielanie się naskórka, jest heterogenną grupą genetycznie uwarunkowanych chorób skóry. Ich wspólną cechą patogenetyczną są mutacje w genach kodujących białka strukturalne odpowiedzialne za adhezję pomiędzy naskórkiem a skórą właściwą. Prowadzą one do nadmiernej kruchości skóry i powstawania pęcherzy nawet po minimalnym urazie. Choroba charakteryzuje się bardzo zróżnicowanym przebiegiem klinicznym, od postaci łagodnych o lokalnym zasięgu do ciężkich, uogólnionych, zagrażających życiu.
Choroba jest spowodowana mutacjami genetycznymi prowadzącymi do dysfunkcji lub całkowitego braku kluczowych białek adhezyjnych, takich jak: keratyny, lamininy, kolagenu typu XVII czy kolagenu typu VII, które warunkują integralność połączenia skórno-naskórkowego. Mutacje te dziedziczone są w sposób autosomalny dominujący lub recesywny, a w części przypadków mogą powstawać spontanicznie (de novo). Bezpośrednią konsekwencją tych defektów molekularnych jest strukturalna niestabilność skóry, objawiająca się jej patologiczną kruchością.