Toczeń rumieniowaty układowy (ICD-10: L93) – autoimmunologiczna choroba tkanki łącznej. Objawy i diagnostyka
Dowiedz się więcej

Zespół Ehlersa-Danlosa jest grupą najczęściej występujących dziedzicznych chorób tkanki łącznej obejmujących kilkanaście podtypów choroby różniących się podłożem molekularnym i przebiegiem klinicznym. Głównymi objawami choroby są nadmierna rozciągliwość skóry, kruchość tkanek i nadmierna ruchliwość stawów. Rozpoznanie zespołu Ehlersa-Danlosa jest bardzo trudne ze względu na złożony obraz kliniczny i brak specyficznych testów genetycznych, wynikający z dużej heterogenności molekularnej. W artykule charakteryzujemy przyczyny i objawy zespołu Ehlersa-Danlosa oraz odpowiadamy na pytanie czy choroba ta jest dziedziczna.

Zespół Ehlersa-Danlosa (EDS) to heterogenna grupa dziedzicznych, genetycznych chorób tkanki łącznej związanych przede wszystkim z nieprawidłową budową kolagenu. Objawy choroby dotyczą głównie skóry, stawów, kości, naczyń krwionośnych i narządów wewnętrznych. Spektrum kliniczne choroby jest dosyć szerokie i może obejmować łagodne zmiany skórne lub nawet nieprawidłowości w funkcjonowaniu narządu ruchu prowadzące do niepełnosprawności. Obecnie wyodrębniono kilka podtypów zespołu Ehlersa – Danlosa, a za najczęściej występujące uznaje się typ hipermobilny, naczyniowy i klasyczny. Pozostałe typy choroby występują z niską częstotliwością.
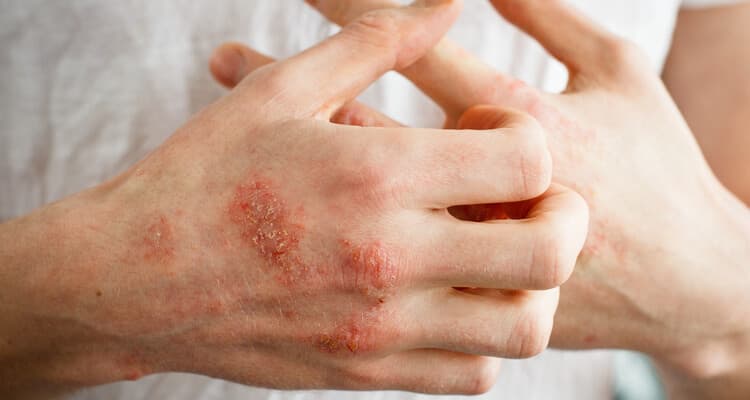
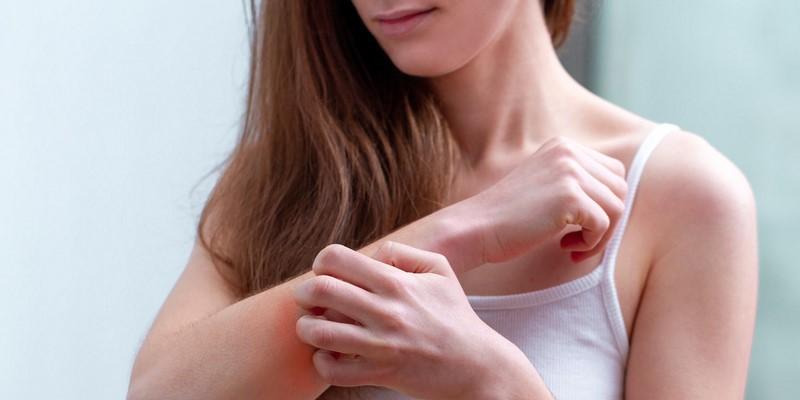
Charakterystycznym objawem choroby jest nadmierna rozciągliwość skóry. Tylko w typie naczyniowym zespołu Ehlersa – Danlosa skóra jest przezroczysta i cienka z uwidocznionymi żyłami podskórnymi i niską zawartością podskórnej tkanki tłuszczowej. Skóra u pacjentów jest bardzo delikatna narażona na uszkodzenia. Obserwuje się pękanie skóry w okolicach łokci czy kolan. Gojenie się ran jest utrudnione a powstające blizny są duże. Na skórze mogą pojawić się liczne krwiaki, przekrwienia i wybroczyny.
U chorych na EDS obserwuje się nadmierną ruchliwość większości stawów (wyjątek typ naczyniowy). Nasilenie nadmiernej hipermobilności stawów jest różne i zależy od wieku i płci. Nadmierna ruchliwość stawów u dzieci może prowadzić do licznych urazów, spowolnionego rozwoju motorycznego, utrudnionego chodzenia, wysięku w stawach i nadmiernego bólu kończyn.
U pacjentów z typem naczyniowym EDS charakterystyczne są powikłania sercowo-naczyniowe, manifestujące się jako rozwarstwienia tętnic, żylaki, przetoki tętniczo-żylne, tętniaki i samoistne pęknięcia tętnic. U większości pacjentów pojawiają się również zaburzenia funkcjonowania układu pokarmowego.
Ze względu na mutacje genów warunkujących syntezę i prawidłowe funkcjonowanie kolagenu, tkanka łączna pacjentów z EDS może być słaba i delikatna w różnych narządach takich jak płuca, pęcherz moczowy, jelita czy dno tętnicy. Pacjenci odczuwają problemy ze wzrokiem m.in. astygmatyzm, szybko postępujące wady wzroku (krótkowzroczność, dalekowzroczność), rozwarstwienie siatkówki. Mogą pojawić się problemy psychiatryczne takie jak depresja, nerwica, zaburzenia snu.
Chorzy mają liczne problemy stomatologiczne związane z łatwym uszkodzeniem śluzówki jamy ustnej, osłabieniem szkliwa, paradontozą, złamaniami zębów i zaburzenia immunologiczne związane ze zwiększonym ryzykiem wystąpienia alergii/uczuleń. Obserwuje się również wysoki odsetek objawów neurologicznych związanych z przewlekłym bólem, chronicznym zmęczeniem i zaburzeniami zmysłu orientacji ułożenia części własnego ciała.
Częstotliwość występowania zespołu Ehlersa-Danlosa jest różna i zależy od podtypu choroby. Najczęściej diagnozowana jest postać klasyczna która występuje u około 1 pacjenta na 20 000 urodzeń. Postać naczyniowa choroby, która związana jest z najcięższym przebiegiem i najgorszym rokowaniem występuję z częstością około 1:100 000.
Przyczyną choroby są mutacje genowe dla genów kolagenu, wywołujące nieprawidłowości w procesie syntezy i obróbki potranslacyjnej kolagenu, nieprawidłową biosyntezę glikozoaminoglikanów oraz zaburzenia procesów wewnątrzkomórkowych. Diagnostyka zespołu Ehlersa-Danlosa opiera się głównie na analizie obrazu klinicznego oraz wykluczenia występowania innych chorób tkanki łącznej.
Kolagen jest jednym z głównych składników tkanki łącznej i stanowi jedną trzecią całkowitego białka organizmu ludzkiego. Kolagen odpowiedzialny jest za prawidłową elastyczność i napięcie skóry. Cząsteczkami składowymi kolagenu są aminokwasy – przede wszystkim prolina, hydroksyprolina, glicyna i hydroksylizyna. Struktura kolagenu może zmieniać się pod wpływem działania czynników zewnętrznych. Białko to buduje mięśnie, kości, skórę i ścięgna, charakteryzuje się dużą wytrzymałością. Budowa i struktura kolagenu zależy od miejsca jego występowania oraz funkcji którą pełni.
Głównym zadaniem kolagenu jest funkcja budulcowa, oznacza to, że stanowi jeden z podstawowych materiałów strukturalnych dla wielu narządów, pełni również funkcję ochronną dla narządów wewnętrznych. Kolagen wpływa na przyspieszenie gojenia się ran i powstawania blizn. Białko to może wiązać cząsteczki wody, zapewniając tym samym odpowiednie nawodnienie skóry. Niestety, wraz z wiekiem synteza wytwarzanego w organizmie kolagenu drastycznie spada. Wolne rodniki, nadmierne opalanie się, za wysokie lub za niskie temperatury oraz czynniki chorobotwórcze mogą również przyspieszyć degradację włókien kolagenu.
Spadek zawartości kolagenu w organizmie może prowadzić do pogorszenia kondycji skóry, problemów ze stawami i ścięgnami jak również trudnościami w poruszaniu się. Zbyt mała ilość kolagenu może prowadzić również do wypadania włosów, ponieważ kolagen odpowiedzialny jest za dostarczanie cebulkom włosowym aminokwasów niezbędnych do odpowiedniego wzrostu. Endogenna synteza kolagenu zmniejsza się po upływie 25 roku życia i przybiera na sile w okresie przekwitania u kobiet ze względu na zmiany hormonalne. Kolagen uczestniczy również w prawidłowym funkcjonowaniu układu kostnego.
Niedobory tego białka mogą wpływać na zwiększenie częstotliwości występowania złamań. Kolagen uczestniczy w produkcji mazi stawowej oraz wpływa na poprawę funkcjonalności chrząstki stawowej, zapewniając jej odpowiedni kształt i odporność na rozciąganie. Kolagen uczestniczy również w regulacji funkcjonowania układu immunologicznego – ograniczając rozpowszechnianie się w organizmie drobnoustrojów chorobotwórczych. Obecnie odkryto ponad 30 różnych typów kolagenu, których wytwarzanie związane jest z odpowiednim genem. Mutacje tych genów mogą wpłynąć na zburzenie syntezy i struktury kolagenu, co skutkuje wystąpieniem chorób genetycznych m.in. takich jak zespół Ehlersa- Danlosa.
Zespół Ehlersa-Danlosa dziedziczony jest głównie autosomalnie dominująco, jednak niektóre typy EDS mogą dziedziczyć się autosomalnie recesywnie. W przypadku dziedziczenia autosomalnego dominującego wystarczy tylko jedna kopia uszkodzonego genu aby wystąpiła choroba, więc nosiciele danej mutacji będą osobami chorymi, manifestującymi objawy choroby. Prawdopodobieństwo przekazania choroby potomstwu, gdy jeden z rodziców jest chory, w tym przypadku wynosi 50%.
Dziedziczenie autosomalne recesywne oznacza że do wystąpienia objawów choroby niezbędne jest odziedziczenie dwóch kopii wadliwego genu. Jeżeli jeden z rodziców jest chory a drugi zdrowy i nie posiada wadliwej kopii genu, to u potomstwa nie powinny wystąpić objawy choroby, ale dzieci będą nosicielami choroby. Część mutacji warunkujących wystąpienie zespołu Ehlersa-Danlosa powstaje de novo – oznacza to, że wcześniej mutacja ta nie występowała u rodziców i powstała spontanicznie dopiero u dziecka.
Obecnie nie istnieje leczenie przyczynowe zespołu Ehlersa-Danlosa. Współczesna terapia skupia się głównie na łagodzeniu objawów choroby. Pacjenci powinni korzystać z rehabilitacji, aby zwiększyć ich siłę mięśniową a tym samym zmniejszyć ryzyko zwichnięć i urazów stawów. Bóle stawów i kończyn można leczyć lekami przeciwbólowymi. Pacjenci powinni unikać ciężkiej pracy fizycznej i intensywnych treningów siłowych. Chorzy podlegają systematycznej kontroli kardiologicznej.