Łuszczyca krostkowa – jak wygląda? Przyczyny i leczenie
Dowiedz się więcej

Gronkowcowy zespół oparzonej skóry, znany w literaturze medycznej również jako choroba Rittera jest ciężką, zagrażającą życiu chorobą dermatologiczną wywołaną przez toksynotwórcze szczepy bakterii Staphylococcus aureus. Zespół ten charakteryzuje się rozległym złuszczaniem naskórka, które klinicznie przypomina głębokie oparzenia termiczne. Nazwa jednostki chorobowej wywodzi się od nazwiska niemieckiego pediatry, Gottfrieda Rittera von Rittershaina. To on w 1878 roku jako pierwszy opisał tę przypadłość u niemowląt, choć już wcześniej podobne obserwacje poczynili inni klinicyści. Współcześnie, dla odróżnienia od toksycznej martwicy naskórka (zespół Lyella) o podłożu polekowym, w piśmiennictwie anglojęzycznym powszechnie stosuje się akronim SSSS (Staphylococcal Scalded Skin Syndrome).
SSSS nie jest typową inwazyjną infekcją tkanki skórnej, lecz zatruciem, tzw. toksykozą. Przyczyną jest bakteria S. aureus, produkująca egzotoksyny złuszczające, zwane eksfoliatynami (ETA i ETB). Natomiast tylko około 5% wszystkich szczepów S. aureus posiada zdolność do produkcji tych toksyn. Zmiany skórne nie są bezpośrednim skutkiem nacieku bakteryjnego w naskórku. Powstające pęcherze i złuszczające obszary są jałowe. Toksyny, produkowane w odległym ognisku zakażenia (np. w nosogardzieli, uchu środkowym, spojówkach, pępku lub w zakażonej ranie), przenikają do krwiobiegu i rozsiewają się drogą krwionośną po całym organizmie, i to właśnie one są przyczyną rozwijającej się choroby Rittera.
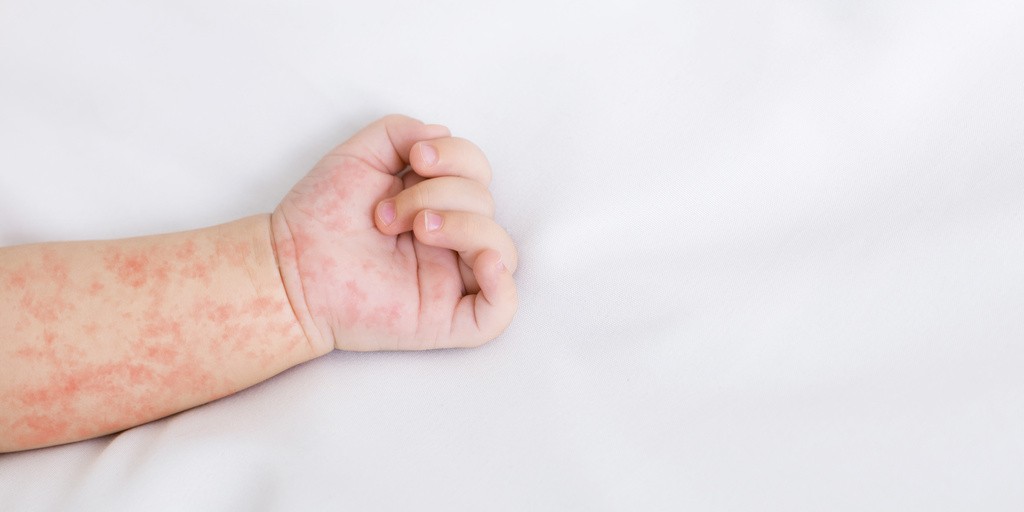
Choroba Rittera występuje stosunkowo rzadko. Szacuje się, że na milion dzieci ta jednostka chorobowa rozwinie się u około 8 pacjentów, podczas gdy u dorosłych jest to zaledwie 1 przypadek na milion.
Predyspozycja populacji pediatrycznej do rozwoju SSSS wynika z dwóch głównych czynników fizjologicznych. Po pierwsze, niedojrzałość mechanizmów odpornościowych, a po drugie niedojrzałość czynnościowa nerek. Eksfoliatyny są wydalane przez nerki, a u małych dzieci klirens nerkowy jest znacząco obniżony, co prowadzi do dłuższej obecności toksyny w surowicy. U dorosłych, choroba rozwija się niemal wyłącznie u pacjentów z ciężkimi deficytami odporności (np. chorzy po przeszczepach, z nowotworami układu krwiotwórczego, zakażeniem HIV) lub z przewlekłą niewydolnością nerek. Śmiertelność w tej grupie jest dramatycznie wysoka i sięga 50-60%, podczas gdy u dzieci leczonych wynosi około 1-5%.
Diagnoza SSSS opiera się przede wszystkim na obrazie klinicznym. Przebieg choroby jest charakterystyczny i gwałtowny. Okres prodromalny trwa od 24 do 48 godzin. Dominują w nim objawy nieswoiste, przypominające ciężkie zakażenie górnych dróg oddechowych:
Następnie pojawia się uogólniona, delikatna wysypka rumieniowa, przypominająca płonicę, która jest bolesna przy dotyku. W ciągu kolejnych 24-48 godzin dochodzi do tworzenia się dużych, wiotkich pęcherzy, które wypełnione są jałowym, przejrzystym płynem. Pęcherze te pękają pod wpływem minimalnego urazu lub ucisku. Po pęknięciu pęcherzy odsłania się wilgotna, sącząca, jaskrawoczerwona powierzchnia, która sprawia wrażenie oparzonej. Złuszczanie przebiega wielkimi płatami. Charakterystyczną cechą różnicującą SSSS od innych ciężkich dermatoz pęcherzowych jest całkowite oszczędzenie błon śluzowych, czyli jama ustna, gardło, spojówki i okolice narządów płciowych pozostają niezmienione. Wokół naturalnych otworów ciała tj. usta czy nos, mogą tworzyć się promieniste, powierzchowne szczeliny i strupy.
Celem diagnostyki mikrobiologicznej jest identyfikacja ogniska pierwotnego, które może być zlokalizowane w nosogardzieli (wymaz z nosa i gardła), spojówkach, krwi (posiew krwi) lub wokół pępka u noworodków. Badanie płynu z pęcherzy nie ma żadnej wartości diagnostycznej, ponieważ jest on jałowy, co odróżnia SSSS od liszajca pęcherzowego, gdzie zmiany są typowo ropne i bogate w bakterie.
Postępowanie diagnostyczne powinno obejmować:
Różnicowanie w procesie diagnostycznym powinno obejmować przede wszystkim toksyczną martwicę naskórka (zespół Lyella), liszajec pęcherzowy oraz chorobę Kawasaki.
Podstawą jest eradykacja szczepu S. aureus w celu zatrzymania produkcji toksyny. Antybiotykoterapia musi być wdrożona niezwłocznie, empirycznie, przed otrzymaniem wyników posiewów.
Leczenie farmakologiczne opiera się na:
Postępowanie wspomagające to przede wszystkim opieka nad uszkodzoną skórą. Ponieważ dochodzi do ogromnych strat płynów przez barierę naskórkową, kluczowe jest dożylne nawadnianie i ścisłe monitorowanie gospodarki elektrolitowej. Skóra wymaga delikatnej pielęgnacji: stosuje się jałowe, nieprzywierające opatrunki, maści osłaniające np. z wazeliną oraz wanny z emolientami. Pacjenci wymagają silnej analgezji, ponieważ zmiany są bardzo bolesne.
Przy szybkim wdrożeniu właściwego leczenia, dzieci z SSSS zwykle wracają do pełni zdrowia w ciągu 5-10 dni. Blizny nie powstają, ponieważ pękanie pęcherzy następuje na bardzo powierzchownym poziomie. Powikłania obejmują wtórne zakażenia w tym posocznicę, zapalenie płuc, ale również odwodnienie i zaburzenia termoregulacji. U dorosłych, ze względu na współistniejące choroby podstawowe, rokowanie jest niepomyślne, a zgon często następuje w wyniku sepsy lub niewydolności wielonarządowej, a nie bezpośrednio zmian skórnych.
Gronkowcowy zespół oparzonej skóry (choroba Rittera) stanowi nagły stan, który mimo rzadkiego występowania wymaga błyskawicznej interwencji diagnostycznej i terapeutycznej. Kluczową rolę w patogenezie odgrywają egzotoksyny Staphylococcus aureus, a nie sam naciek bakteryjny. Szybkie działanie w tym przypadku ratuje życie i pozwala uniknąć trwałych blizn, ponieważ naskórek regeneruje się całkowicie.
Mgr Kinga Dworak
Choroba Rittera objawia się gwałtownym początkiem z wysoką gorączką, a następnie w ciągu 24-48 godzin pojawia się bolesny, uogólniony rumień oraz duże, wiotkie pęcherze, które łatwo pękają. Prowadzi to do rozległego złuszczania naskórka wielkimi płatami, odsłaniając wilgotną, czerwoną powierzchnię przypominającą oparzenie, z całkowitym oszczędzeniem błon śluzowych.
Sam zespół nie jest zaraźliwy, ponieważ jest on wynikiem działania toksyny krążącej we krwi chorego. Jednak bakteria Staphylococcus aureus, która wywołuje chorobę, jest zaraźliwa - może przenosić się drogą kropelkową lub przez bezpośredni kontakt z nosicielem, ale u zdrowych osób wywołuje zwykle jedynie łagodne zakażenia, a nie ciężką postać zespołu.
Przy szybkim wdrożeniu właściwego leczenia (dożylne antybiotyki i opieka wspomagająca) dzieci z SSSS zwykle wracają do pełnego zdrowia w ciągu 5 do 10 dni. Samo złuszczanie ustaje po około 2-3 dniach od rozpoczęcia antybiotykoterapii, a pełna regeneracja naskórka trwa zwykle około 5-7 dni.

